Protein folding and AlphaFold explained for software engineers
You might be wondering what's stopping us from building a simulation of a complex living organism from DNA. Researching this question might take forever, but it’s reasonable to start with one of the first roadblocks - the protein-folding problem.
Protein folding is one of the most important unsolved fundamental problems in biology. Solving it would significantly speed up disease research, drug discovery, and production of synthetic proteins. Examples of diseases caused by misfolded proteins are: Alzheimer's, Parkinson’s, Huntington's, and even several allergies.
Protein folding can be an especially interesting challenge for engineers and data scientists since the state-of-the-art software (Alpha Fold) is developed by one of the most famous AI companies - DeepMind. And they are not experts in biology; their expertise lies in machine learning.
Introduction into Protein Folding
Let's start with the basics: protein is a large biomolecule and it consists of chains of amino acids (also known as amino acid residues). In the software input, you will see a string similar to “QETRKKCTEMKKKFKNCE…”.
But just knowing such a sequence of amino acids is not enough to understand what the protein encoded by such sequence will do in a body, because the function of a protein highly depends on its three-dimensional shape.
For example, antibody proteins have a Y shape that forms hooks and latches onto viruses.
So, to understand what sequences of amino acids are actually doing in the body we need to be able to predict a protein structure from a sequence.
In other words, we want to fold a protein into a 3-dimensional shape from its amino acids.
Determining just a single protein structure in a lab can cost up to hundreds of thousands of dollars and might take years.
Machine Learning and CASP
Machine learning is a good fit for the protein-folding problem for several reasons:
- It's a complex problem that is hard to model and understand.
- It has complex local and long-range interactions.
- The rules of physics for making a protein crystal are unknown.
- It has experimental data - about 150,000 solved structures. Unfortunately, they are highly redundant
- The problem seems to be amenable to some kind of human intuition.
- It is impossible to randomly enumerate through all possible structures of a molecule due to an astronomical number of potential shapes.
- The goals are well-defined.
Critical Assessment of protein Structure Prediction (CASP), is an experiment for protein structure prediction that takes place every two years. During the last CASP 13 experiment in 2018, AlphaFold outperformed all other models by a big margin.
It should be noted that CASP measures results for 2 domains, one of them is "Free Modeling" and it means that the protein structure is completely new. And in "Template-based modeling", which means that the protein structure is very similar to an existing known "solved protein". AlphaFold was experimenting only in the "Free Modeling" category and created high-accuracy structures for 24 out of 43 free modeling domains. The next best model achieved only 14.
Neural network
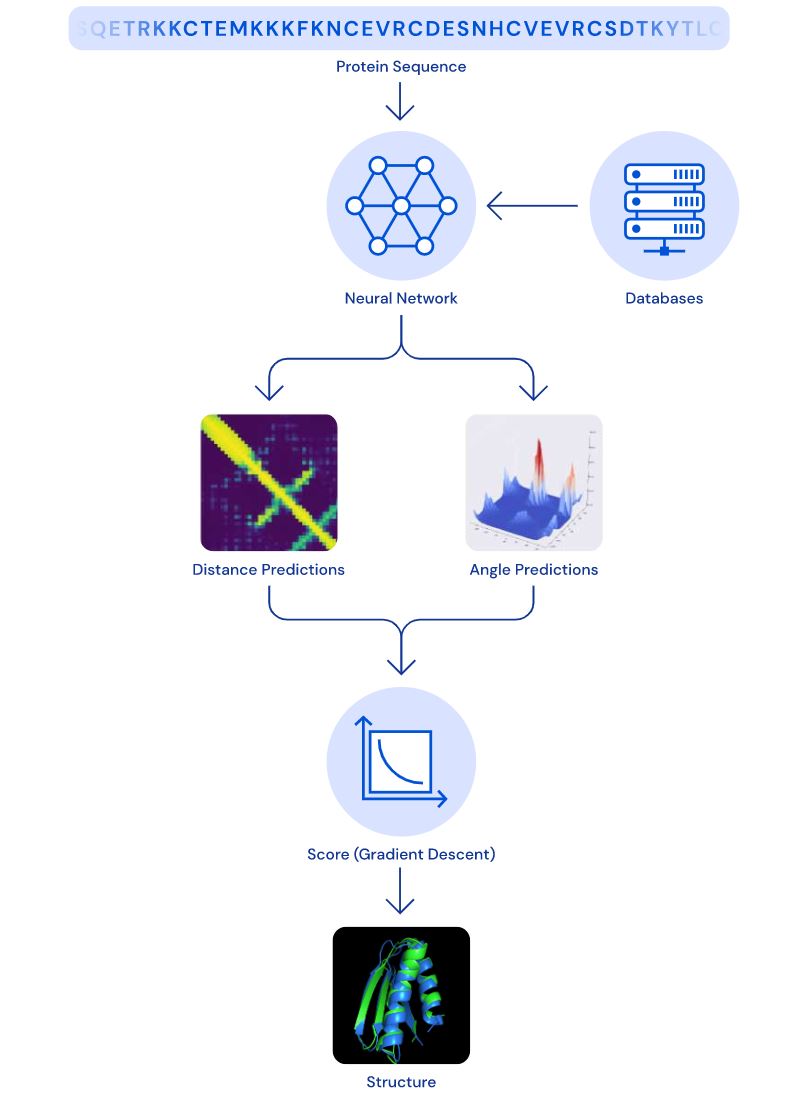
A few words about the input first. Proteins are evolved from common ancestors and because of that will have a lot of commonalities. That means that we can take the source protein sequence and compare it to known proteins to extract “coevolutionary” features. The accuracy of the result highly depends on the number of coevolutionary features.
The first neural network takes previously coevolutionary features as an input and produces a large number of distributional predictions for distances between pairs of amino acids and angles between chemical bonds that connect those amino acids. Based on distance predictions, it’s possible to build a heatmap such as can be seen in the picture below.
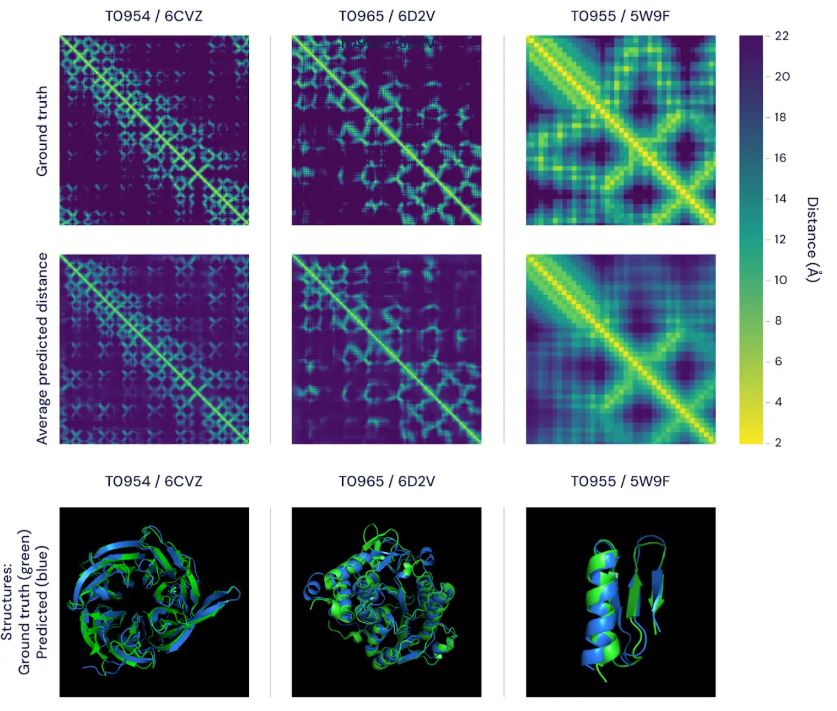
All we need to know is whether residues are connected or not. However, making distance predictions is a very important idea that allowed AlphaFold to make connectivity predictions with higher accuracy.
Cropping is another interesting technique. AlphaFold is working and was trained on 64x64 regions of distance matrices, so bigger proteins are comprised of more than 1 region.
This cropping method has the following advantages:
- Data augmentation. Thanks to crop offsets randomization Deepmind managed to significantly augment the training dataset.
- Helps with distributed training
- Constraints memory usage
- Helps to avoid overfitting
- 64 is a good matrix size because it’s big enough to model long-range interactions
Another technique that Deepmind used is searching for known proteins that have similar pieces and replacing fragments of a predicted protein structure with a known piece.
The neural network for distance and angle prediction has 21 million parameters. Compare it to a recently released GPT-3 with 175 billion parameters and you can realize how relatively small it is. The good news is Deepmind reported that their learning curve wasn’t flattening and AlphaFold kept improving with more parameters.
And then there is a second subsystem that works on the whole protein. Based on distance and angle distributional predictions of the previously described network, AlphaFold uses gradient descent together with some geometry and math to output the final protein structure predictions.
For more details, read the original paper.
For more details, read the original paper.
Implications
The current state of AlphaFold doesn't produce good enough results to be used in practice. But if we extrapolate the progress of the last 2 CASPs then in 4 years protein folding problem might get solved. Unfortunately, there is no guarantee and before the last 2 CASP competitions, there was almost no progress between each experiment for 10 years.
One of the ways to keep improving protein folding networks is to produce more data. Although there are more and more solved proteins each year, the growth rate is nowhere near other datasets that are used for image recognition, for instance.
There are projects that try to motivate people to help with creating a big enough database of solved protein structures, e.g. Folding@Home.
Speculations
Outside of AlphaFold, the Deepmind company is working on creating an Artificial General Intelligence. And since they were acquired by Google their algorithms are used in Android and Play Market.
They are not experts in anything related to biology, that's why I find it very interesting that Deepmind can just come into the new field with academic researchers that have been working on the problem for many years and beat all of them by a huge margin. And it's not just purely academics, some of the competing labs are sponsored by huge corporations such as Roche.
Mohammed AlQuraishi suggested that this might have happened because pharmaceutical companies don't invest enough in research or invest in research that is so narrow, that it barely contributes to the understanding of basic biology and rarely attracts talent. Mohammed also thinks that these companies don't invest enough into ML right now.
References
- Deepmind’s article in the Nature magazine - “Improved protein structure prediction using potentials from deep learning”
- Blog post by Deepmind - AlphaFold: Using AI for scientific discovery
- Github - AlphaFold repository
- How Biology and living cells "compute". Conference talk oriented for software engineers - Youtube
Comments
Post a Comment